La preuve définitive que l'ARNm/LNP est bel et bien une THÉRAPIE GÉNÉTIQUE.

https://sashalatypova.substack.com/p/make-your-dollar-bills-even-crispr?
Rendez vos billets de banque encore plus CRISPR ! ( crisp = croustillant)
Sasha Latypova
J'aborderai sous peu le sujet principal de mon article, le succès fulgurant de CRISPR (une chance sur un million). Pour bien le comprendre, je vous invite à lire l'article de Lies are Unbecoming :

Je travaillais sur cet article depuis quelques semaines lorsque je suis tombée sur l'histoire de cet enfant dont la vie a été brisée par les vaccins. J'ai le cœur brisé pour tous les enfants victimes d'effets indésirables liés à la vaccination et pour leurs parents. Cependant, cette histoire en particulier est absolument bouleversante :
Cachée au cœur de la biographie d'Andrew Webster parue en 2023, « The Wolf You Feed » , l'entraîneur de rugby à XIII le plus titré d'Australie livre une confession si crue, si bouleversante, qu'elle remet en question tout ce que l'on nous dit sur les effets indésirables des vaccins infantiles et les maladies génétiques. Bennett, un homme qui a passé un demi-siècle à perfectionner l'art de dissimuler ses émotions, a révélé que son fils Justin avait subi des lésions cérébrales suite à une vaccination de routine à l'âge de quatre mois. La véritable tragédie ne réside pas seulement dans ce qui est arrivé à Justin Bennett en 1977, mais aussi dans la manière dont un cas manifeste d'empoisonnement par vaccin a été rétrospectivement requalifié en une maladie génétique appelée syndrome de Dravet, déplaçant ainsi la responsabilité de l'injection qui a détruit le cerveau d'un nourrisson vers les parents qui auraient soi-disant transmis des gènes défectueux.
Plus de 40 ans après, les victimes ont été de nouveau ridiculisées en prétendant qu'une « mutation génétique rare » en était responsable :
Le fardeau psychologique que représentent les diagnostics génétiques pour les parents est incommensurable. Wayne et Trish Bennett sont passés de la certitude que leur fils avait subi un préjudice vaccinal – un dommage externe infligé à leur enfant en bonne santé – à l'annonce qu'ils avaient transmis des gènes défectueux qui condamnaient Justin dès sa conception. Il ne s'agit pas simplement d'un changement de classification médicale ; c'est une redéfinition fondamentale de la responsabilité morale. Le coupable devient le destin, le crime devient l'hérédité, et les parents deviennent des complices involontaires de la destruction de leur enfant.
Comme l'auteur le souligne à juste titre, les « maladies génétiques » constituent une autre supercherie scientifique, où les effets indésirables des vaccins et autres dommages iatrogènes sont reconditionnés :
Le témoignage personnel de Wayne Bennett concernant les effets indésirables de la vaccination de son fils trouve un appui scientifique considérable dans l'analyse exhaustive de Jonathan Latham et Allison Wilson, intitulée « The Great DNA Data Deficit ». Leurs recherches fournissent les bases empiriques permettant de comprendre comment le déterminisme génétique est devenu ce que l'on pourrait décrire comme « une fraude à au moins 70 %, voire jusqu'à 90 % » – un édifice frauduleux servant de couverture à des empoisonnements de masse et à la pollution industrielle.
Justin Bennett a été intoxiqué par le vaccin DTP à l'âge de quatre mois, ce qui a provoqué des crises d'épilepsie ayant entraîné des lésions cérébrales irréversibles. Pendant plus de quarante ans, sa famille a dû lui prodiguer des soins constants. En 2019, ses parents ont été accusés de leur avoir transmis des gènes défectueux responsables d'une « épilepsie génétique rare/syndrome de Dravet », dont la prévalence, selon certains généticiens, serait de 1 sur 15 000 à 1 sur 40 000.
C'était en 2019.
En 2025, une cabale de sorcières se faisant appeler l'Hôpital pour enfants de Philadelphie (CHOP) a lancé une offensive dévastatrice. Voici « Bébé KJ », atteint d'une maladie génétique parmi les plus rares qui soient… Passons maintenant au cœur du sujet :
La thérapie CRISPR personnalisée du « bébé KJ » pour une « maladie génétique ultra-rare »
Si vous ne savez pas ce que signifie CRISPR, vous pouvez lire les inepties de science-fiction à ce sujet sur Wikipédia. Ou demandez à votre « expert en génomique » préféré du mouvement « liberté en matière de santé », et il vous en parlera pendant des heures. Ce Substack ne diffuse pas de pseudo-science. Mon avis personnel et éclairé sur CRISPR : c'est du poison dans des nanoparticules lipidiques. Je parlerai plus bas de la mixture spécifique du petit KJ, je vous le promets : c'est une arnaque absolument passionnante.
Comme l'ont rapporté avec enthousiasme l'industrie pharmaceutique et la presse scientifique en mai dernier : un nourrisson connu publiquement sous le nom de KJ (KJ Muldoon) est devenu le premier patient à recevoir une thérapie génique CRISPR in vivo personnalisée pour traiter une maladie métabolique ultra-rare et potentiellement mortelle (un déficit sévère en CPS1 – carbamoyl phosphate synthétase 1). Ce cas et les détails cliniques ont été publiés dans un article évalué par des pairs et dans des communiqués institutionnels en mai 2025.
Le déficit en CPS1 est un trouble du cycle de l'urée qui empêche le foie d'éliminer l'ammoniaque produite lors du métabolisme des protéines ; non traité, il provoque une hyperammoniémie dangereuse pouvant entraîner des lésions neurologiques ou le décès. La prise en charge standard repose souvent sur une restriction stricte des protéines alimentaires, des médicaments chélateurs d'azote et, dans les cas les plus graves, une transplantation hépatique.
Quelles sont les chances qu'un bébé naisse avec cette « maladie génétique ultra-rare » ? Une sur 1 300 000, affirme la science sans ambages. Une sur 1,3 million ! Incroyable, la génétique ! Une chance sur 15 000 était déjà alarmante. Et ce chiffre est totalement fictif, bien sûr. Mais ces Australiens étaient des amateurs. Avec l'équipe multidisciplinaire CHOP, on entre dans la cour des grands. Cette équipe comptait des noms prestigieux issus non seulement des plus grandes universités, mais aussi d'entreprises comme Danaher, Acuitas (propriétaire de la plateforme d'armement LNP) et, bien sûr, AmplifyBio, la société de J.D. Vance !
J'avais un pressentiment et j'ai donc consulté les données du VAERS. J'ai recherché un seul symptôme de la supposée « maladie génétique ultra-rare » du bébé KJ : une augmentation du taux d'ammoniaque dans le sang. J'ai trouvé environ 80 cas, dont la moitié concernaient des nourrissons et de jeunes enfants après une vaccination contre le rotavirus et le TDP (celui qui a paralysé Justin Bennett en Australie), et l'autre moitié des adultes après une vaccination contre la COVID-19. Les cas présentaient un large éventail de lésions hépatiques, allant d'une élévation des marqueurs hépatiques à l'hépatite, en passant par les convulsions et les lésions cérébrales permanentes, comme celles de Justin Bennett.
L'équipe du CHOP a bien sûr imputé ce qui est très probablement une lésion vaccinale (soit le vaccin DTP du bébé KJ, soit la vaccination de la mère pendant la grossesse) à une mutation « ultra-rare » et l'a élégamment imputée au fait que le père de KJ serait un « mutant génétique » :
L'équipe a choisi de cibler la mutation Q335X (de Dad), qui nécessitait de reconvertir un codon stop en un acide aminé fonctionnel en changeant une adénine (A) en une guanine (G).
Ils se sont alors lancés dans la création d'un jus injectable miracle qu'ils ont baptisé « kayjayguran abengcemeran, ou k-abe ». Personnellement, je trouve que ça ressemble beaucoup trop à « kayfabe » dans le monde du catch. Voici la description du produit tirée des 4 500 pages (!!! Je ne plaisante pas) de l'article du NEJM, ainsi que le protocole complet que j'ai achetés. Merci aux abonnés payants de me fournir des lectures aussi fascinantes avant de dormir, de quoi me donner des cauchemars fonctionnels !
Le médicament (MD) est une thérapie d'édition de base encapsulée dans des nanoparticules lipidiques (NPL) ciblant le foie. Il comprend une substance médicamenteuse (SM) à base d'ARN messager (ARNm) codant pour un éditeur de base adénine (EBA), une SM à base d'ARN guide (ARNg) unique qui se lie à l'EBA une fois exprimé dans les cellules cibles, et des excipients lipidiques constituant le système de délivrance des SM LNP pour les SM d'acides nucléiques. Le MD sera administré par voie intraveineuse.
Attendez… reprenons… quoi ? L’ARNm dans les LNP est une THÉRAPIE GÉNÉTIQUE ??? C’est-à-dire qu’on « corrige » les gènes « incorrects » ultra-rares avec ça ? Et ce n’est pas un vaccin ? Allô, FDA… Bueller… Bueller… Buellerrrrr…
Cette thérapie « CRISPR » est quasiment identique aux vaccins contre la Covid, jusqu'à la substitution de l'uridine et un poids moléculaire similaire :
L'ARNm comprend la coiffe 5', la région non traduite 5' (UTR), la séquence codante ABE, la région non traduite 3' (UTR) et la queue polyadénylée 3'. La séquence codante ABE, longue de 4 818 nucléotides, est optimisée pour les codons par minimisation de l'uridine et présente une substitution de toutes les uridines par le nucléotide modifié N1-méthylpseudouridine.
Passons à la suite… Comment a-t-on créé ce mélange miraculeux de gènes « corrects » ? Voici le processus de fabrication miraculeux illustré à la figure 1 :
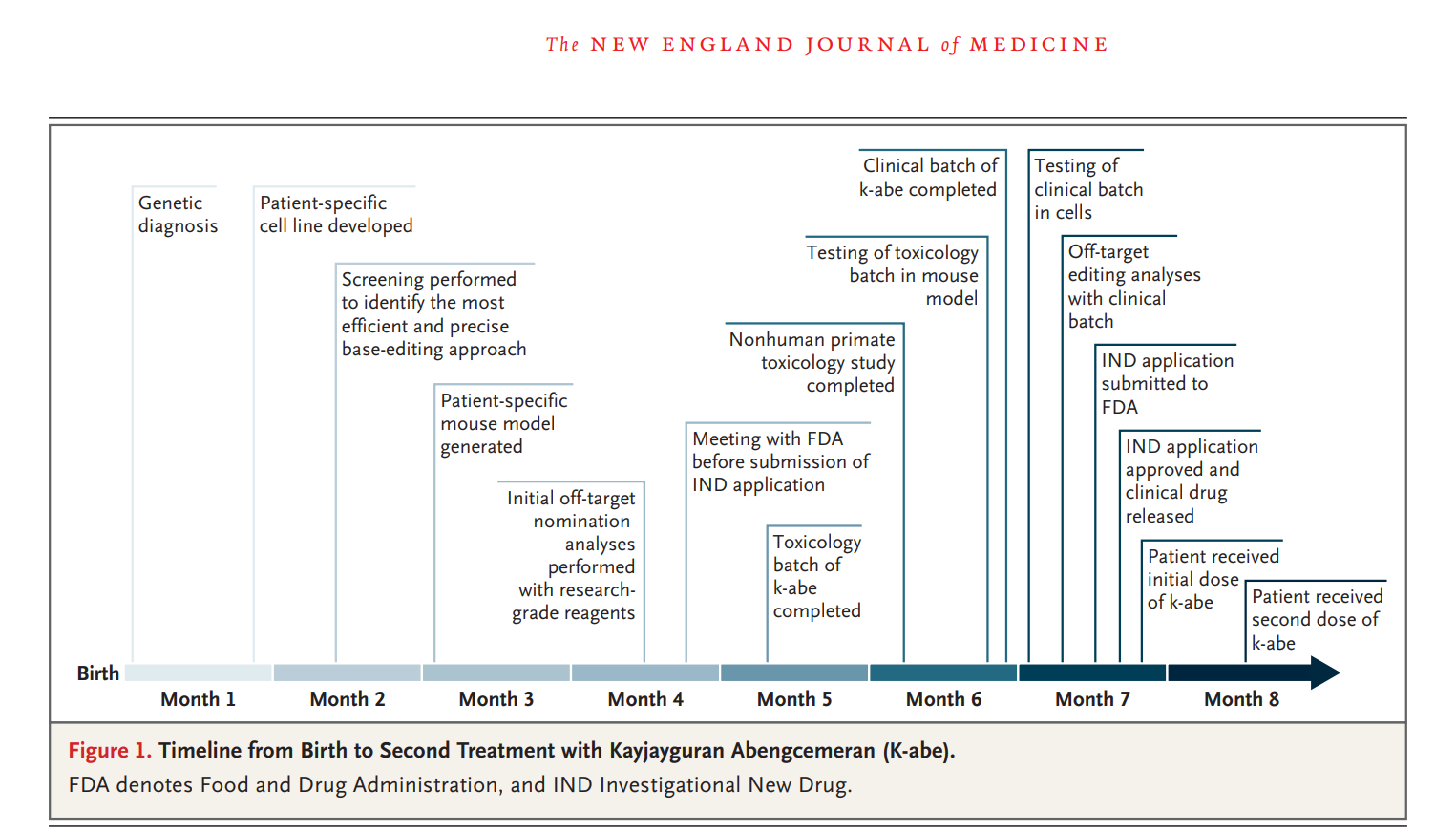
Tout cela paraît extrêmement impressionnant et « réel ». Le vocabulaire est savant, le graphique complexe et hallucinant. Voyez : ils ont utilisé des lignées cellulaires hépatiques humaines (HuH-7) modifiées pour porter la mutation de KJ, puis ils ont « créé des souris génétiquement modifiées » avec « l'erreur génétique exacte » de KJ (!!!). Ils ont même réalisé une analyse hors cible à l'aide de plusieurs outils de pointe comme ONE-seq , CHANGE-seq et GUIDE-seq , pour s'assurer que l'éditeur ne ciblait pas les « mauvaises » séquences d'ADN. Cependant, toute cette pseudo-science n'est que pure fantaisie, sans aucune validation clinique. Ils s'en remettent à un modèle informatique inventé de toutes pièces.
j'ai écrit un très bon article explicatif sur l'absurdité de ce domaine de la « science » :
Le problème que j'ai soulevé dès le départ est le suivant : les analyses PCR et le séquençage génétique se déroulent en milieu liquide ; tous les ingrédients, sous forme de poudre chimique, sont littéralement dissous dans un liquide. Comment, dès lors, peuvent-ils prétendre « lire » une séquence nucléotidique de gauche à droite, comme un objet physique, alors que cet objet est dissous ?
Bon, alors ces mêmes « ciseaux moléculaires intelligents » qui transportent des valises de « colle moléculaire intelligente » savent lire les composés chimiques dans l'eau de gauche à droite (ils sont d'origine occidentale, vous voyez, pas asiatique). Les « oligonucléotides » fabriqués en laboratoire se présentent sous forme de flacons visiblement vides. Il faut CROIRE qu'ils contiennent des gènes « corrects » ! Oh, mais il faut aussi lire en petits caractères que la grande majorité des oligos sont produits hors des normes cGMP, donc le contenu du flacon vide n'est pas forcément le « bon » génétique. De plus, même certifié cGMP, un produit ne peut être certifié « correct » qu'à environ 70 %. La science, c'est passionnant, non ?
KJ a reçu plusieurs perfusions de « jus CRISPR » contenant les « gènes corrects » à partir de l'âge de 6 à 7 mois environ. L'article du NEJM rapporte que ce traitement a permis une meilleure tolérance aux protéines alimentaires, une réduction de la dépendance aux médicaments chélateurs d'azote, et que le nourrisson a pu quitter l'hôpital en bonne santé, sous suivi régulier. L'innocuité à long terme est bien sûr inconnue, et aucune information concernant l'état de santé de KJ n'a été publiée à ce jour.
On n'a pas pratiqué de biopsie du foie, car le nourrisson était « trop fragile ». Mon Dieu, ils lui ont fait subir toutes sortes d'examens, y compris une ponction lombaire, mais pas ça… Pourquoi ? Bien sûr, il faut croire que la « génétique a été corrigée ». Personne ne veut de preuves concrètes, car au fond, tout le monde sait qu'on prie un dieu virtuel et qu'on orchestre une supercherie.
Bon, qui se soucie encore de ce bébé… On ne peut pas l'injecter continuellement avec ce vaccin à ARNm personnalisé dans des LNP (non vaccin, non, non, non !). Pas encore, en tout cas. Pour permettre l'injection continue de ce vaccin à ARNm dans des LNP à de nombreux autres bébés porteurs de gènes incorrects,
CHOPsters vise l'approbation de la FDA !
Selon Endpoint News :
Les scientifiques à l'origine de cette découverte sont désormais prêts à fabriquer davantage de médicaments sur mesure dans le cadre d'un essai clinique qui pourrait débuter l'année prochaine.
S'exprimant mardi lors de la réunion de la Société européenne de thérapie génique et cellulaire en Espagne, Kiran Musunuru, le scientifique de l'Université de Pennsylvanie qui a codirigé le développement de la thérapie de Baby KJ, a déclaré avoir récemment rencontré la FDA pour discuter d'un « protocole directeur » pour un essai de phase 1/2 qui recrutera des patients atteints de l'un des six troubles du cycle de l'urée et pourrait transformer la thérapie unique de Baby KJ en une procédure approuvable et reproductible.
Ce groupe de maladies métaboliques est rare mais bien compris.
Il y a beaucoup à dire sur cette dernière phrase. Tout d'abord, remarquez l'omniprésence du terme « maladie métabolique ». Je l'ai vu tellement souvent que je commence à me demander si Calley « Métabulaire » Means n'approuve pas tous les arguments des laboratoires pharmaceutiques, universitaires et gouvernementaux concernant les produits biologiques. Tout cela pour justifier ces « maladies métaboliques » ultra-rares, mais paradoxalement très répandues.
Je vous conseille, chers lecteurs, de penser « dissimulation des effets indésirables des vaccins » chaque fois que vous entendez un agent du HHS, un larbin de l'industrie pharmaceutique ou une militante de MAHA (quel que soit son genre) s'extasier sur les « maladies métaboliques ».
Ensuite, comment une maladie « ultra-rare » qui touche une personne sur 1,3 million peut-elle être « bien comprise » ? Des études cliniques prospectives ont-elles été menées sur une maladie aussi rare ? Non ! C'est hors de prix et cela prendrait environ 400 ans, compte tenu de sa rareté. Bien sûr, cette « compréhension » provient – vous l’aurez deviné – de prières adressées à un faux dieu informatique et de l’utilisation du petit KJ comme cobaye. Car la nouvelle MAHA-FDA, contrairement à la précédente FDA de Biden, aussi mauvaise soit-elle, veut protéger les singes.
Endpoints News s'est entretenu avec Musunuru (auteur principal du projet de jus CRISPR k-[f]abe) et a publié l'entretien . Cette discussion a révélé « la clémence inattendue de la FDA, qui a quasiment éliminé l'obligation de mener des études animales dans le cadre du prochain essai clinique. Le principal souci n'est pas d'étudier l'innocuité et la toxicologie chez les animaux avant d'injecter de l'ARNm aux nouveau-nés, mais bien de garantir un essai clinique équitable.
Et par « équitable », on entend en réalité : les contribuables doivent payer pour que les médecins du CHOP injectent de l'ARNm aux nouveau-nés, autrement dit, un « remboursement ». »
Critères d'évaluation : En quoi un essai clinique est-il plus équitable ?
Musunuru : Si cinq ou dix patients suffisent pour obtenir une autorisation conditionnelle, l’essai sera rapidement complet. Une fois l’autorisation obtenue, il devient remboursable et donc autofinancé, car vous récupérez au moins votre investissement.
Et lorsqu'on parle de « remboursement » pour les « thérapies géniques », cela peut représenter des millions de dollars par dose !
Objectifs : Outre l’étude sur les troubles du cycle de l’urée, vous travaillez également sur une étude de grande envergure visant à développer des médicaments CRISPR pour plusieurs mutations différentes responsables de la phénylcétonurie (PCU), une autre maladie métabolique. Comment s’est déroulée votre réunion avec la FDA à ce sujet ?
Musunuru : Concernant la phénylcétonurie (PCU), nous avions proposé de réaliser des études toxicologiques sur des primates non humains et des rats pour deux des six variants. Pour les variants trois, quatre, cinq et six, nous avions prévu de nous limiter à des études cellulaires in vitro. La FDA nous a alors fait une petite surprise : les études toxicologiques ne sont nécessaires que pour un seul des six variants, et il peut s’agir soit d’études sur des primates non humains, soit d’études sur des rats, mais pas sur les deux, afin de préserver la santé animale. C’est donc une approche relativement nouvelle.
Et puis — je crois que c'était une concession faite à notre communauté universitaire — ils ont précisé que l'étude toxicologique n'avait pas besoin d'être entièrement conforme aux BPL (bonnes pratiques de laboratoire, une norme utilisée dans le cadre du développement). Elle pouvait s'en approcher, à condition de documenter les différences. Nous avons obtenu tout ce que nous avions demandé, et même plus. Le lendemain de cet échange, nous avons déposé une demande de réunion concernant les troubles du cycle de l'urée.
La porte est ouverte, faisons passer le plus de choses possible.
Exactement. Quelle phrase finale fantastique ! Elle résume tout, n'est-ce pas ? La nouvelle MAHA-FDA veut « épargner les animaux » et n'a plus besoin des BPL, qui sont pourtant obligatoires pour une autorisation de mise sur le marché de produits biologiques (AMM). Mais qui se soucie encore des lois ?
Entre-temps, alors que j'écrivais cet article, j'ai vu passer dans mon fil d'actualité une nouvelle concernant une autre entreprise pionnière dans le domaine de CRISPR, Intellia : un patient participant à un essai clinique d'Intellia CRISPR est décédé après avoir été hospitalisé pour une lésion hépatique .
Un homme d'une quatre-vingtaine d'années, récemment hospitalisé pour une lésion hépatique quelques semaines après avoir reçu une thérapie expérimentale de modification génétique, est décédé mercredi soir.
La cause exacte du décès du patient, annoncée jeudi après-midi par Intellia Therapeutics, n'était pas immédiatement connue, a indiqué la société. La semaine dernière, Intellia avait signalé une élévation de grade 4 de deux biomarqueurs – les transaminases hépatiques et la bilirubine – répondant aux critères de la loi de Hy, un critère clinique indiquant un risque d'atteinte hépatique grave, voire mortelle, d'origine médicamenteuse.
Ce traitement expérimental, appelé nexiguran ziclumeran, utilise des nanoparticules lipidiques pour acheminer les éditeurs de gènes, qui inactivent ensuite le gène codant pour les protéines mal repliées. En fait, il s'agit aussi d'ARNm dans les LNP, pas dans les vaccins !
Ce n'est pas la première fois qu'un décès survient lors d'un essai clinique de thérapie génique, admet la presse à la solde de l'industrie pharmaceutique. On le sait bien, n'est-ce pas ? Des millions de personnes sont mortes ou ont subi de graves préjudices suite à des injections d'ARNm qui ne sont pas des thérapies géniques !
Un homme qui avait bénéficié d'une thérapie de modification épigénétique personnalisée pour sa forme rare de dystrophie musculaire est décédé en 2022 des suites d'une réaction immunitaire au traitement.
Je dois m'arrêter un instant. « Édition épigénétique », c'est vraiment hallucinant ! L'épigénétique est définie comme « l'étude des modifications de l'expression des gènes qui surviennent sans altération de la séquence d'ADN ». En réalité, l'épigénétique est une solution de facilité, car la « génétique » est une absurdité, un culte religieux où l'on vénère un dieu informatique, et qui, de ce fait, ne peut rien expliquer sur l'hérédité, la normalité biologique, l'anormalité, la maladie, etc. L'« épigénétique » à la rescousse ! Comment font-ils pour « modifier » ça… mystère. Enfin, on sait comment : ils ont tué le type…
Mais les scientifiques ont déterminé que c'était la forte dose de vecteurs viraux utilisée pour administrer la thérapie à ses muscles — et non le système CRISPR lui-même — qui était en cause.
…et il a mis ça sur le compte de ses muscles défectueux que la « modification épigénétique » était censée guérir ! Chers lecteurs, je vous jure, je n’ai fait que copier-coller tout ça des experts, je n’invente rien.
Une autre personne est décédée d'un arrêt cardiaque cinq semaines après avoir reçu un médicament hypolipémiant développé par Verve Therapeutics en 2023. Ce décès a été jugé sans lien avec le médicament. Un autre patient participant à un essai clinique a subi un grave infarctus du myocarde le lendemain de l'administration du traitement ; cet infarctus a été jugé « potentiellement lié au traitement ».
Oups ! Je crois qu'il faudrait vérifier que le petit KJ va bien, non ?
Articles précédemment publiés sur CRISPR et les thérapies géniques :




Œuvre d'art du jour : Roses roses, huile sur panneau, 11x14 pouces .


Commentaires
Enregistrer un commentaire