Réduire de 95 % ou 76 % un risque d'infection de 1,5 % avec les vax Pfizer ou Moderna - Lettre de Karen Kingston
De : https://karenkingston.substack.com/p/draft-sept-2-2021-letter-to-fda-commissioner?
ÉBAUCHE - Lettre du 2 septembre 2021 à la commissaire de la FDA, Janet Woodcock, concernant l'approbation de la FDA

On m'a demandé de rédiger une lettre à Janet Woodcock concernant l'approbation par la FDA de COMIRNATY pour un sénateur. Ceci est le projet de lettre que j'ai envoyé le 2 septembre 2021.
2 septembre 2021
Janet Woodcock, M.D.
Commissaire par intérim
Administration des aliments et des médicaments
10903, avenue New Hampshire
Silver Spring, MD 20993
Cher Dr Woodcock :
Le 22 août 2021, je vous ai envoyé une lettre vous demandant de reconsidérer la décision de la Food and Drug Administration (FDA) de renoncer à organiser une réunion du comité consultatif sur les vaccins et les produits biologiques apparentés (VRBPAC) avant d'approuver une demande de licence biologique (BLA) pour le EUA Pfizer-BioNTech ARNm COVID-19 « vaccin ».
Votre réponse écrite du 23 août 2021, ci-après dénommée « RÉPONSE BLA DE LA FDA », n'a pas raisonnablement répondu aux préoccupations concernant l'efficacité, les risques pour la sécurité et les événements indésirables graves (EIG) du vaccin EUA Pfizer-BioNTech COVID-19. . Voici quelques exemples de votre manque de transparence, de crédibilité et d'intégrité dans votre RÉPONSE BLA de la FDA.
RÉPONSE BLA DE LA FDA : ¶1:2
"Cette décision a été prise après un examen et un examen minutieux, notant qu'il n'y avait pas de nouvelles préoccupations de fond concernant la sécurité ou l'efficacité de ce vaccin Pfizer-BioNTech depuis les deux réunions consultatives précédentes au cours desquelles il a été discuté." – Janet Woodcock, M.D.
LA FDA AFFIRME « AUCUNE NOUVELLE PRÉOCCUPATION DE FOND CONCERNANT LA SÉCURITÉ OU L'EFFICACITÉ DU VACCIN EUA PFIZER-BIONTECH COVD-19 »
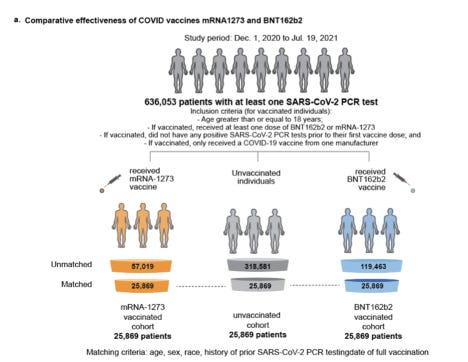
Avant votre réponse écrite du 23 août et après la tenue de la dernière réunion du VRBPAC le 10 juin 2021 , le 9 août 2021, la clinique Mayo a publié une analyse rétrospective bien conçue de 636 053 patients, âgés de 16 ans ou plus, dans 5 États, des sujets qui ont TOUS reçu des tests PCR entre le 1er décembre 2020 et le 19 juillet 2021. Les sujets ont reçu soit le « vaccin » EUA Pfizer-BioNTech (BNT161b), le « vaccin » EUA Moderna (ARNm-1273), ou aucune intervention médicale.
Les enquêteurs ont fait un travail méticuleux pour faire correspondre exactement les membres des groupes d'étude de la triple cohorte par sexe, race, origine ethnique, état de résidence et fréquence des antécédents de test PCR.
Les objectifs de l'étude de la Mayo Clinic étaient de déterminer les résultats suivants de l'efficacité du vaccin (EV) et de l'efficacité du vaccin pour chaque cohorte :
Efficacité du vaccin (EV) = réduction de l'IRR SARS-CoV-2 Infection* (+PCR)
· *VE est le résultat principal de tous les vaccins COVID-19
· Voir page 15, ¶11, Pfizer IND, Phase 1/2/3, Vaccins COVID-19 à base d'ARN
Résultats d'efficacité des vaccins
· Hospitalisation associée au COVID-19 - dans les 21 jours suivant l'infection = (+PCR)
· Admission aux soins intensifs associée au COVID-19 - dans les 21 jours suivant l'infection = (+PCR)
· Mortalité associée au COVID-19 - dans les 28 jours suivant l'infection = (+PCR)
· Infection soudaine = + PCR confirmant l'infection par le SRAS-CoV-2 survenant au moins 14 jours après la deuxième dose d'ARNm1273 ou de BNT162
Les enquêteurs de l'étude Mayo Clinic ont fourni des détails spécifiques sur la cohorte du Minnesota, produisant 3 cohortes de 25 689 sujets chacune.
EFFICACITÉ DES VACCINS
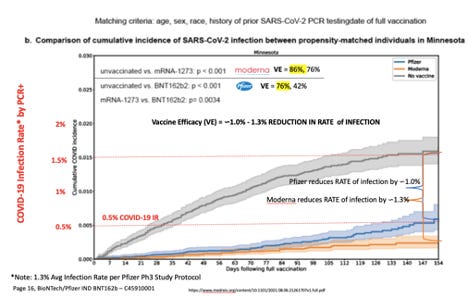
En ce qui concerne «l'efficacité du vaccin» (VE), les «vaccins» Pfizer-BioNTech (BNT162b) et Moderna (ARNm-1273) ont pu réduire le taux d'infection (IRR) d'environ 1% chacun par rapport à l'absence d'intervention médicale à tout. Bien que les données de la Mayo Clinic montrent que Moderna a été initialement 86 % plus efficace que le placebo pour réduire le risque d'infection de 1 % par rapport à "l'efficacité du vaccin" de 76 % de Pfizer, aucun de ces points de données n'a de bénéfice clinique significatif pour prévenir l'infection ou le propagation du virus SARS-CoV-2. Sur une période de quelques mois, le «taux de réussite de Moderna à ne rien faire d'intérêt clinique» est tombé à 76% et celui de Pfizer à 42%.
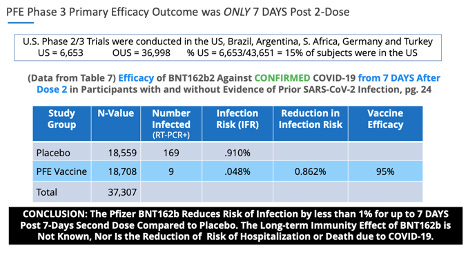
Les points de données VE étaient similaires dans les essais de phase 3 de Pfizer. Le principal critère d'évaluation de Pfizer était une « efficacité du vaccin » de 95 % pour réduire le risque d'infection de moins de 1 % (0,86 %) 7 jours après la deuxième dose.
Dr Woodcock, vous avez des décennies d'expérience pour déterminer si un médicament ou un vaccin s'avère offrir une efficacité de «signification clinique» et pas seulement de «signification statistique», ainsi que l'importance de communiquer des revendications de sécurité et d'efficacité claires et précises au peuple américain sous 21 USC Sec. 502 – Allégations fausses ou trompeuses.
Selon la page 16, ¶1, Pfizer IND, Phase 1/2/3, RNA-Based COVID-19 Vaccines , le risque de taux d'infection pour le SRAS-CoV-2 est de 1,3 %.
AVANTAGES DE L'EFFICACITÉ DES VACCINS (VE) - QUESTIONS :
1. Selon votre opinion d'expert, pensez-vous qu'une chance de 95 % de réduire le risque d'infection par le SRAS-CoV-2 de 1,3 % à environ 1,287 % présente un avantage clinique significatif pour les sujets de l'étude et le peuple américain ? Oui ou Non. Veuillez expliquer votre réponse.
2. Vous affirmez qu'il n'y a eu "aucune nouvelle préoccupation de fond concernant la sécurité et l'efficacité du BNT162b depuis la réunion du 10 juin 2021 du VRBPAC". Pourquoi n'avez-vous pas trouvé inquiétant que dans une triple cohorte de plus de 75 000 patients COVID-19, 1 % des sujets injectés avec le vaccin Pfizer-BioNTech aient subi des effets indésirables graves ?
3. Pourquoi la FDA n'a-t-elle pas commandé les données complètes de l'ensemble Mayo Clinical en envoyant un e-mail à venky@nference.net pour examen et analyse avant de fournir l'approbation BLA ?
4. les données de la Mayo Clinic, pensez-vous qu'une chance de 86 % ou 76 % de réduire le risque d'infection par le SRAS-CoV-2 de 1,5 % à environ 1,48 % pendant jusqu'à deux semaines après la deuxième dose est un bénéfice clinique significatif pour les sujets d'étude et le peuple américain ? Oui ou Non. Veuillez expliquer votre réponse.
5. En tant que citoyen américain et contribuable, croyez-vous à l'avantage clinique de réduire le risque d'infection de 1 % pour une maladie qui a un taux d'infection extrêmement faible de 1,3 % et un taux de mortalité inférieur à 0,01 % pour les personnes en bonne santé , vaut les quelque 100 milliards de dollars dépensés pour commercialiser ces vaccins « COVID-19 » et des centaines de milliards de dollars de plus pour les rappels et les systèmes de surveillance des vaccins, au détriment d'autres initiatives de soins de santé qui peuvent promouvoir la forme physique, la nutrition , bien-être émotionnel, habitudes de sommeil saines, prévenir la toxicomanie et l'alcoolisme, prévenir l'obésité, réduire l'intimidation et enseigner des comportements sains en ligne, et d'autres programmes qui peuvent aider les Américains et leurs enfants à mener une vie plus épanouissante, joyeuse et réussie ? Oui ou Non. Veuillez expliquer votre réponse.
6. Les plus de 100 milliards de dollars dépensés et qui continuent d'être dépensés pour les vaccins COVID-19 sont-ils bien investis dans l'intérêt supérieur de la santé et du bien-être physiques et émotionnels du peuple américain et de ses enfants ? Oui ou Non. Veuillez expliquer votre réponse.
Sachant que la norme dans les communications d'informations médicales est de présumer que la compréhension d'une allégation de produit ou d'un message éducatif est par une personne sans expertise médicale ou scientifique avec un niveau de lecture de 6e année, vous êtes pleinement conscient que lorsqu'un sujet voit "95 % efficace », ils comprennent que cela signifie une réduction de 95 % du risque absolu d'infection par rapport au placebo ou au groupe témoin. Il est trompeur de prétendre supposer que l'Américain moyen croirait que «95%» efficace signifie 95% de chances d'avoir une réduction de 1% du risque absolu d'infection.
Vous trouverez ci-dessous un exemple du CDC affirmant que le Pfizer-BioNTech est efficace à 95 % pour prévenir une infection confirmée par, vraisemblablement, le SRAS-CoV-2, le virus qui cause le COVID-19.

RÉCLAMATIONS VE - QUESTIONS
7. Pourquoi autorisez-vous le CDC à faire des déclarations fausses et trompeuses concernant l'efficacité des vaccins au peuple américain ?
8. Pourquoi n'avez-vous pas demandé au CDC de fournir une déclaration précise concernant TOUTE l'efficacité absolue du vaccin ? Selon les données déposées auprès de la FDA, ils sont efficaces à environ 1 % pour réduire le taux d'infection par le SRAS-CoV-2, PAS 86 %, 92 %, 95 % ou 100 % ou tout autre chiffre faux et trompeur que le CDC continue de fournir et propager avec la permission et les conseils de la FDA.
EFFICACITÉ DES VACCINS
En plus de mesurer «l'efficacité du vaccin», la clinique Mayo a également mesuré plusieurs résultats d'efficacité du vaccin, y compris les «infections soudaines COVID-19». L'infection post-vaccinale a été définie comme le SRAS-CoV-2 confirmé par un test PCR positif survenant au moins 14 jours après la deuxième dose d'ARNm1273 ou de BNT162 (c'est-à-dire des sujets «entièrement vaccinés»).
Selon les tableaux 1 et 8 de l'étude, il y avait 21 179 sujets entièrement vaccinés dans le groupe Moderna (ARNm-1273), dont 106 ont subi des « infections soudaines par le COVID-19 » = 0,5 %. Il y avait 22 064 sujets entièrement vaccinés dans le groupe Pfizer BioNTech (BNT162b), dont 220 ont connu des «infections majeures au COVID-19» = 1,0%.
« INFECTIONS SOUDAINES (PERCÉES) PAR LA COVID-19 »
Vous trouverez ci-dessous le tableau 8 de l'étude détaillant la liste principale des symptômes des « infections post- vaccinales au COVID-19 » chez les personnes entièrement vaccinées. L'incidence post-vaccinale (révolutionnaire) de 1 % dans le groupe Pfizer des cas de COVID-19 a entraîné des maladies graves telles que l'ARD/ALD (souvent causée par l'immunogénicité ou l'ADE), une lésion/maladie rénale aiguë, l'anémie (alias thrombocytopénie), la thrombose, l'arrêt cardiaque, syndrome de fatigue chronique (principalement causé par une encéphalite et/ou une myélite), insuffisance cardiaque (causée par une inflammation de l'HVG, alias myocardite), coagulation intravasculaire disséminée, septicémie (alias MIS-C), accident vasculaire cérébral et al. Cela était alarmant pour les enquêteurs en ce qui concerne l'efficacité des vaccins pour prévenir une infection grave et renforce potentiellement le besoin de rappels.

Je ne crois pas que les maladies graves indiquées dans la liste des « INFECTIONS SOUDAINES PAR LE COVID-19 » de la clinique Mayo soient dues au coronavirus. Je crois qu'il s'agissait d'événements indésirables graves causés par les « vaccins » COVID-19 eux-mêmes. Le VRBPAC de la FDA s'est réuni le 22 octobre 2020 et a dressé une liste des effets indésirables graves prévisibles (EIG) des VACCINS COVID-19 de l'EUA . Bien que je ne sois ni médecin ni scientifique, je suis alphabétisé et la liste des « INFECTIONS COVID-19 » de la Mayo Clinic correspond textuellement (avec quelques modifications mineures) à la liste des SAE prévisibles des vaccins COVID-19 selon la réunion du VRBPAC.

Dr Woodcock, je ne crois pas que vous vous soyez fourvoyée ou que vous manquiez d'intelligence pour comprendre que les preuves cliniques prouvent de manière écrasante que les vaccins Pfizer-BioNTech et Moderna COVID-19 provoquent ces événements indésirables graves, à un taux minimum de 1% dans un période de quelques jours ou quelques mois seulement après la deuxième dose. Il est important de noter que le tableau 8 n'inclut pas les « infections aiguës » ou les hospitalisations après une dose.
À ce jour, il y a eu un total d'environ 645 000 Américains qui sont morts avec COVID-19 sur une population d'environ 331,5 mm. Environ 0,2 % d'Américains sont décédés avec le COVDI-19 au cours des 18 derniers mois. En élargissant on peut dire qu' environ 96 750 personnes sont décédées du COVID-19, soit environ 0,03 % au cours des 18 derniers mois.
INFECTIONS SOUDAINES (PERCÉES) /EIG – QUESTIONS :
1. Pourquoi la liste des maladies dans l'étude de la clinique Mayo définies comme des « infections majeures au COVID-19 » correspond-elle presque textuellement à la liste des événements indésirables graves prévisibles créée par le FDA VRBPAC des événements indésirables graves possibles ?
2. Veuillez également expliquer le rapport risque-bénéfice d'être injecté avec le vaccin Pfizer- BioNTech COVID-19 - avec un risque de sécurité de 1 % pour un SAE, et un bénéfice d'efficacité d'une réduction de 1 % du risque d'infection par une maladie avec un taux d'infection de 1,3 % et un taux de mortalité de 0,01 % ou moins pour la majorité des Américains. Veuillez expliciter votre analyse.
3. Pour mieux comprendre la demande tyrannique d'obligations de vaccination dans nos écoles, nos forces armées et nos lieux de travail, veuillez expliquer l'analyse risques-avantages pour que les jeunes adultes et adolescents en bonne santé reçoivent le vaccin Pfizer BioNTech. Veuillez montrer votre analyse.
4. D'après votre analyse, si les jeunes adultes, adolescents et enfants en bonne santé ont moins de 0,004 % de décès dus au COVID-19, pourquoi doivent-ils être vaccinés ?
RÉPONSE BLA DE LA FDA : ¶1:3
"Les informations fournies dans la demande de licence de produits biologiques répondaient à toutes les normes de la FDA en matière de qualité, de sécurité et d'efficacité pour l'homologation." – Janet Woodcock, M.D.
RÉCLAMATION DE LA FDA : "LES INFORMATIONS RESPECTENT TOUTES LES NORMES DE LA FDA EN MATIÈRE DE QUALITÉ, DE SÉCURITÉ ET D'EFFICACITÉ "
Bien que vous indiquiez que les données soumises pour l'approbation de la BLA respectaient toutes les normes de la FDA en matière de qualité, de sécurité et d'efficacité pour la licence, selon 21 USC Sec. 312.44 IND TERMINATION, votre déclaration est malhonnête. Les vaccins EUA COVID-19 n'auraient jamais dû passer les essais humains de phase 1, sans parler des essais de phase 2 ou 3, et obtenir l'approbation complète de l'autorisation. Si vous pensez que je me trompe, veuillez fournir votre contre-argument avec des preuves cliniques documentées pour étayer votre position.
21 USC Sec. 312.44 RÉSILIATION
a) Général. Cette section décrit les procédures selon lesquelles la FDA peut résilier un IND. Si une IND est résiliée, le promoteur doit mettre fin à toutes les investigations cliniques menées dans le cadre de l'IND et rappeler ou autrement prévoir l'élimination de toutes les fournitures inutilisées du médicament. Une action de résiliation peut être fondée sur des lacunes dans l'IND ou dans la conduite d'une enquête en vertu d'un IND. Sauf dans les cas prévus au paragraphe (d) de cette section, une résiliation doit être précédée d'une proposition de résiliation par la FDA et d'une opportunité pour le promoteur de répondre. En général, la FDA n'initiera une action en vertu de cette section qu'après avoir d'abord tenté de résoudre les différences de manière informelle ou, le cas échéant, par le biais des procédures de suspension clinique décrites au § 312.42.
(b) Motifs de résiliation - (1) Phase 1. La FDA peut proposer de résilier une IND au cours de la Phase 1 si elle constate que :
(i) Les sujets humains seraient exposés à un risque déraisonnable et significatif de maladie ou de blessure.
· Selon la propre présentation Powerpoint VRBPAC de la FDA, diapositive 16, datée du 22 octobre 2020, la FDA répertorie les événements indésirables graves anticipés pouvant entraîner des dommages permanents, une invalidité et la mort.
· Par pages 67-68, Sec. 8.3.5.1. Exposition pendant la grossesse (EDP ) : Pfizer IND, Phase 1/2/3, RNA-Based COVID-19 Vaccins et EDP se produit si :
o Un membre féminin de la famille ou un prestataire de soins de santé signale qu'elle est enceinte après avoir été exposée à l'intervention de l'étude par inhalation ou contact cutané.
o Un membre masculin de la famille ou un prestataire de soins de santé qui a été exposé à l'intervention de l'étude par inhalation ou contact cutané expose ensuite sa partenaire féminine avant ou autour de la conception.
o DERNIER PARAGRAPHE Page 68 : Les décès néonatals qui surviennent dans le mois suivant la naissance doivent être signalés, sans égard à la causalité, comme des EIG. En outre, les décès de nourrissons sont liés ou peuvent être liés à l'exposition à l'intervention à l'étude.
· Aux pages 69, Sec. 8.3.5.2. Exposition pendant l'allaitement : Pfizer IND, Phase 1/2/3, RNA-Based COVID-19 Vaccins , Une exposition pendant l'allaitement se produit si :
o Une participante s'avère allaiter pendant qu'elle reçoit ou après l'arrêt de l'intervention de l'étude.
o Il s'avère qu'une femme allaite pendant qu'elle est exposée ou a été exposée à l'intervention de l'étude (c'est-à-dire l'exposition environnementale)….membre de la famille ou prestataire de soins de santé qui signale qu'elle allaite après avoir été exposée à l'intervention de l'étude par inhalation ou contact avec la peau .
La FDA était au courant du risque élevé de malformations congénitales et de décès chez les bébés à naître et les nouveau-nés, et a quand même poursuivi les essais Pfizer-BioNTech COVID-19 et EUA.
Vous trouverez ci-dessous un exemple de la mort tragique d'un petit garçon de 5 mois en parfaite santé à la suite de la propre violation délibérée de la FDA de 21 USC 312.44.

DES QUESTIONS:
· Sur la base du risque de transmission des sujets injectés aux femmes enceintes uniquement, pourquoi la FDA n'a-t-elle pas arrêté les essais des vaccins Pfizer-BioNTech COVID-19 ?
· Sur la base des risques connus de préjudice et de décès pour les nouveau-nés et les nourrissons par excrétion de sujets injectés, pourquoi la FDA n'a-t-elle pas arrêté les essais des vaccins Pfizer-BioNTech COVID-19 ?
· Pourquoi la FDA n'a-t-elle pas averti les femmes enceintes et les parents de nouveau-nés et de nourrissons de ces risques ?
(ii) L'IND ne contient pas suffisamment d'informations requises en vertu du § 312.23 pour évaluer la sécurité des sujets des investigations cliniques.
· La majorité des études animales n'ont jamais été initiées ou sont incomplètes. Veuillez fournir un résumé et des ensembles de données complets de toutes les études animales réalisées à ce jour.
· Parmi les autres études animales, la FDA n'a jamais demandé au fabricant de lancer ou de terminer des études sur la descendance et l'excrétion conformément au document d'orientation de la FDA d'août 2015, " Conception et analyse des études d'excrétion pour la thérapie génique basée sur les virus ou les bactéries et les produits oncolytiques ", ce qui a entraîné des dizaines de millions d'Américains produisent des particules de virion nocives, c'est-à-dire des protéines de pointe, tout en exposant les autres à un risque élevé d'infection par excrétion. Les femmes enceintes sont particulièrement exposées à des risques tératogènes et les nourrissons sont exposés à un risque élevé de maladie grave de la part de membres de la famille et d'amis vaccinés.
· Le risque de préjudice causé par l'excrétion aux femmes enceintes et à leurs nourrissons est évident dans la propre lettre BLA de la FDA datée du 23 août 2021, exigeant d'évaluer ces risques comme indiqué ci-dessus à la page 9, point 10. "Pfizer BioNTech COVID- 19 Exposition pendant la grossesse : une étude non interventionnelle sur l'innocuité post-approbation des résultats de la grossesse et du nourrisson dans l'Organisation des spécialistes de l'information en tératologie (OTIS)/MotherToBaby Pregnancy Registry. »
· Des milliers de femmes enceintes ont fait des fausses couches et de nombreux parents ont perdu leur nouveau-né ou leur nourrisson à cause de la violation délibérée de la FDA de 21 USC 312.44.
· « INSÉRER LES DONNÉES VAERS SUR LES FAUSSES COUCHES ET LES DÉCÈS DE NOUVEAU-NÉS »
DES QUESTIONS:
· Pourquoi la FDA n'a-t-elle pas demandé au CDC, au NIH, aux HSH, aux employeurs, aux gouvernements locaux et fédéraux et aux conseils scolaires d'avertir les mères enceintes et les parents de nouveau-nés et de nourrissons de ces risques ?
· Pourquoi la FDA a-t-elle permis à ces décès de se produire sans intervention et stratégies d'atténuation des risques ?
· Pouvez-vous préciser votre connaissance et votre détermination des risques d'immunogénicité (stimulation dépendante des anticorps), de tératogénicité, d'effets indésirables cardiaques graves, d'effets indésirables cardiovasculaires graves, d'effets indésirables graves touchant le système nerveux central, d'effets indésirables pulmonaires graves, de risques de progéniture ? et l'excrétion virale par rapport aux avantages d'efficacité selon le SARS-Cov-2 et/ou COVID-19 IRR, IFR, CFR et le risque d'hospitalisation par groupe d'âge et comorbidités ?
(iii) Les méthodes, les installations et les contrôles utilisés pour la fabrication, le traitement et l'emballage du médicament expérimental sont inadéquats pour établir et maintenir des normes appropriées d'identité, de concentration, de qualité et de pureté nécessaires à la sécurité des sujets.
· Conformément aux réglementations de l'EUA, la majorité des cGMP (bonnes pratiques de fabrication des consommateurs) ont été abandonnées, y compris l'inspection des installations de fabrication, les laboratoires de test de contrôle de la qualité et la confirmation du contenu final et de la qualité des ingrédients finaux du flacon de vaccin.
· Conformément à la propre lettre BLA de la FDA, le contenu final des flacons de vaccin doit être confirmé d'ici le 6 septembre 2021, ainsi que la finalisation de l'étiquetage, des ingrédients, des effets secondaires et des événements indésirables graves.
QUESTION : Quels PROTOCOLES DE CONTRÔLE ONT ÉTÉ APPLIQUÉS PAR LA FDA POUR GARANTIR des normes appropriées d'identité, de force, de qualité et de pureté nécessaires à la SÉCURITÉ DES SUJETS qui ont été injectés dans le cadre de l'EUA ?
(iv) Les investigations cliniques sont menées d'une manière substantiellement différente de celle décrite dans les protocoles soumis dans l'IND .
· Selon la page 9 de l'IND, Justification : Pfizer IND, Phase 1/2/3, RNA-Based COVID-19 Vaccines , le document indique :
o Les 2 candidats vaccins SARS-CoV-2 qui seront testés dans cette étude sont donc :
§ BNT162b1 (variante RBP020.3) : un modARN codant pour le RBD ;
§ BNT162b2 (variante RBP020.2) : un modRNA codant P2 S.
o Tous les candidats sont formulés dans la même composition de nanoparticules lipidiques (LNP).
o Cette étude vise à étudier l'innocuité, l'immunogénicité et l'efficacité de ces vaccins prophylactiques BNT162 contre le COVID-19.
Le Dr Fauci et de nombreux leaders et influenceurs de la santé ont informé le peuple américain que les vaccins à ARNm produiraient la protéine de pointe du SRAS-CoV-2, le virus qui cause le COVID-19, puis que leur corps produirait des anticorps neutralisants. Cependant, la variante BNT162b2 du vaccin à ARNm ne produit pas la protéine de pointe SARS-CoV-2. BNT162b2 produit la protéine de pointe SP-2 à laquelle 2 prolines ont été ajoutées, modifiant la conformation (forme) et les domaines de liaison au récepteur de la protéine de pointe. Selon une publication du 19 février 2020 dans Science, la protéine de pointe SP-2 produite par les vaccins à ARNm se lie efficacement aux sites récepteurs ACE-2 dans le cœur, les poumons et les reins, entraînant une inflammation, une maladie et/ou la mort, MAIS n'a pratiquement aucune activité de liaison aux anticorps neutralisants de la protéine de pointe SARS-CoV-2.
DES QUESTIONS:
· Selon les données présentées dans l'article du 19 février 2020 dans Science, l'ARNm chimérique à base de coronavirus dans BNT162b produit SP-2, une protéine de pointe très puissante pour induire l'inflammation et la maladie, mais qui n'a aucune activité de liaison aux anticorps neutralisants de la protéine de pointe SARS-CoV-2. À votre avis d'expert, comment le SP-2 produit-il une protection immunitaire contre le SRAS-CoV-2 ?
· Les données sur la conformation et le mécanisme d'action du SP-2 dans Science étaient à votre disposition des mois avant l'approbation de l'IND et l'autorisation de l'EUA du BNT162b. Quelle est la justification clinique de votre approbation de l'IND et de l'EUA pour le BNT162b ?
La page 1, ¶4 de la lettre d'approbation de la FDA du 23 août 2021 indique : "L'examen de ce produit a été associé aux numéros d'essais cliniques nationaux (NCT) suivants : NCT04368728 et NCT04380701."
· NCT04368728 - https://clinicaltrials.gov/ct2/show/NCT04368728
· NCT04380701 - https://clinicaltrials.gov/ct2/show/NCT04368728
Selon les descriptions de l'étude NCT déposées le 30 avril 2020, une troisième version des vaccins Pfizer-BioNTech COVID-19 connue sous le nom de BNT162b2VOC (ou BNT162bSA) serait testée sur les participants à l'essai de phase 3 pour la variante sud-africaine.
En janvier 2021, l'Organisation mondiale de la santé a publié des articles sur l'émergence de variantes du coronavirus, y compris celles d'Afrique du Sud, mais aucune variante sud-africaine n'avait été clairement identifiée en janvier 2021 .
DES QUESTIONS:
· En avril 2020, pourquoi avez-vous approuvé la recherche humaine sur des citoyens américains pour un agent génétique d'ARNm à base de virus chimérique (BNT162bSA) qui n'avait JAMAIS été TESTÉ SUR DES ANIMAUX et pour prétendument prévenir l'infection par un virus qui n'existait même pas encore ?
· Pourquoi avez-vous délibérément violé 21 USC Sec. 312.44 (iv) ? Pourquoi n'avez-vous pas immédiatement mis fin à la demande de même seulement commencer les essais de cet agent biologique ?
Par page, la lettre BLA du 23 août et les lettres d'autorisation EUA de la FDA, l'autorisation et l'approbation par la FDA des vaccins Pfizer BioNTech COVD-19 sont associées au (x) produit(s) utilisé(s) dans les essais IND/NCT qui incluent les versions d'ARNm suivantes et différentes dosages;
1. BNT162b1 : dose de 10 µg : l'ARNm code pour les domaines de liaison aux récepteurs RBD
2. BNT162b2 : dose de 10 µg : l'ARNm code pour PS 2 - protéine de pointe
3. BNT162b1 : dose de 20 µg : l'ARNm code pour RBD - les domaines de liaison aux récepteurs
4. BNT162b2 : dose de 20 µg : l'ARNm code pour PS 2 - protéine de pointe
5. BNT162b1 : dose de 30 µg : l'ARNm code pour RBD - les domaines de liaison aux récepteurs
6. BNT162b2 : dose de 30 µg : l'ARNm code pour PS 2 - protéine de pointe
7. BNT162b2SA : dose de 30 µg : l'ARNm code pour la protéine de pointe sud-africaine
8. BNT162b1 : dose de 100 µg : l'ARNm code pour RBD - les domaines de liaison aux récepteurs
9. BNT162b2 : dose de rappel de 5 µg : l'ARNm code pour PS 2 - protéine de pointe
10. BNT162b2 : dose de rappel de 10 µg : l'ARNm code pour PS 2 - protéine de pointe
11. BNT162b2 : dose de rappel de 30 µg : l'ARNm code pour PS 2 - protéine de pointe
12. Autre : Placebo
DES QUESTIONS:
· Quel est le mécanisme d'action de l'ARNm codant pour les domaines de liaison au récepteur du 162BNTb1 dans la prévention de l'infection par le SARS-CoV-2, le virus qui cause le COVID-19 ?
· Existe-t-il des attributs pathogènes connus de l'ARNm BNT162b1 ?
Les Américains ont l'impression que s'ils ont reçu le vaccin EUA Pfizer ou recevront le vaccin COMIRNATY BLA APPROVED, les deux vaccins sont la version Phase 3 BNT162b2 à une dose de 30 mcg par dose. Sur la base de la formulation confuse des lettres EUA et BLA, cela n'est pas clair. Précisez s'il vous plaît.
DES QUESTIONS:
· Quelle(s) version(s) d'ARNm se trouvaient dans les vaccins EUA Pfizer-BioNTech administrés au peuple américain ? A quelle(s) dose(s) ?
· Quelles mesures de contrôle de la qualité étaient en place pour garantir que les vaccins BNT162b2 fabriqués dans le cadre de l'EUA (ou d'autres versions), avant l'approbation COMIRNATY BLA, étaient conformes aux ingrédients répertoriés dans la NDA de phase 3 déposée le 20 novembre 2020 ?
· Quelle(s) formulation(s) de BNT162b est/sont dans le COMIRNATY sous licence FDA ?
· Quel(s) dosage(s) d'ARNm est/sont dans COMIRNATY ?
(v) Le médicament est promu ou distribué à des fins commerciales non justifiées par les exigences de l'enquête ou autorisées par le § 312.7.
· L'article 312.7 concerne les enquêteurs qui ont soumis des données non fiables. Les Américains comptent 85 % des enquêteurs et leurs sujets se trouvaient en dehors des États-Unis, au Brésil, en Argentine, en Afrique du Sud, en Allemagne et en Turquie.
DES QUESTIONS:
· Quels conseils de surveillance américains indépendants étaient en place pour garantir la fiabilité des données soumises ?
· Quels chercheurs et/ou leurs sites de recherche clinique avaient des liens financiers avec les fabricants (Pfizer-BioNTech) et des parties ou organisations gouvernementales non américaines ? Quels étaient ces liens financiers ?
En ce qui concerne les données non fiables, p. 24, tableau 7. de la phase 3 de Pfizer, 20 novembre 2020, soumission EUA , il y avait 169 sujets placebo qui présentaient des symptômes de COVID-19 confirmés par un test PCR positif, et seulement 9 sujets dans le groupe BNT162b, ce qui a entraîné la ' statistiquement significative' 'efficacité du vaccin.'
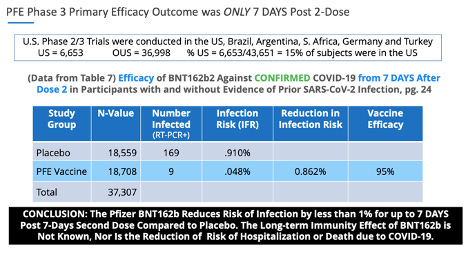
Fait intéressant, p. 41, ¶ 2: 2 de la phase 3 de Pfizer, 20 novembre 2020, soumission de l'EUA , indique clairement: «Les cas suspects de COVID-19 survenus dans les 7 jours après toute vaccination étaient de 409 dans le groupe vacciné contre 287 dans le groupe placebo .”
Si ces données avaient été prises en compte de manière précise et éthique, le principal critère d'évaluation de l'« efficacité du vaccin » pour le vaccin Pfizer-BioNTech contre la COVID-19 aurait montré une signification statistique nulle.
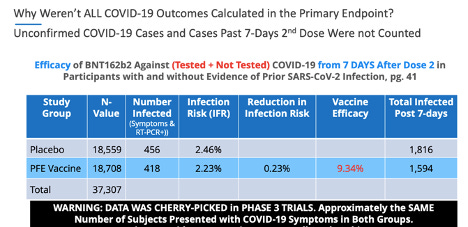
La page 41, ¶2: 3 du document Pfizer poursuit en déclarant: «Il est possible que le déséquilibre des cas suspects de COVID-19 survenant dans les 7 jours suivant la vaccination représente la réactogénicité au vaccin avec des symptômes qui chevauchent ceux de COVID-19. Dans l' ensemble, cependant, ces données ne soulèvent pas d'inquiétude quant au fait que la notification spécifiée par le protocole des cas suspects, mais non confirmés de COVID-19 pourrait avoir masqué des événements indésirables cliniquement significatifs qui n'auraient pas été détectés autrement.
· Par § 312.7.e) Si le commissaire de la FDA détermine, après que les données non fiables soumises par l'investigateur ont été éliminées, que la poursuite de l'approbation du produit pour lequel les données ont été soumises ne peut être justifiée, le commissaire de la FDA procédera au retrait de l'approbation du produit conformément aux dispositions applicables des statuts pertinents.
À mon avis, votre négligence grave en ignorant ces violations flagrantes du § 312.7.e ne peut s'expliquer que par une faute intentionnelle. Cependant, je vais vous donner l'occasion de répondre, car je pense que le peuple américain a le droit de connaître les motivations qui sous-tendent vos décisions.
DES QUESTIONS:
· En tant que commissaire de la FDA, pourquoi n'avez-vous pas souligné que les données fournies pour Pfizer-BioNTech ne sont pas fiables de l'aveu même du sponsor ?
· En tant que commissaire de la FDA, pourquoi avez-vous permis au promoteur de faire des affirmations contradictoires selon lesquelles les maladies graves qui se sont manifestées dans la semaine suivant la deuxième dose de BNTA162b chez 409 sujets supplémentaires N'ÉTAIENT NI DES CAS DE COVID-19 ni de ÉVÈNEMENTS GRAVES NÉFASTES ?
(vi) L'IND, ou tout amendement ou rapport à l'IND, contient une déclaration fausse d'un fait important ou omet des informations importantes requises par cette partie.
· DÉCLARATION FAUX DE FAIT MATÉRIEL : Les produits BNT162b utilisés dans les essais ne répondent pas à la définition clinique, selon la FDA, ou à la définition légale, selon l'USPTO, d'un vaccin. Les produits BNT162b répondent à la définition d'une "thérapie génique virale "
· Selon le propre document d'orientation de la FDA d'août 2015, " Conception et analyse des études d'excrétion pour la thérapie génique basée sur des virus ou des bactéries et les produits oncolytiques ", "Les produits de thérapie génique sont tous les produits qui médient leurs effets par la transcription et/ou la traduction du matériel génétique transféré et/ou en s'intégrant dans le génome de l'hôte et qui sont administrés sous forme d'acides nucléiques (ARNm), de virus ou de micro-organismes génétiquement modifiés.
(vii) Le promoteur omet d'enquêter et d'informer rapidement la Food and Drug Administration et tous les enquêteurs des effets indésirables graves et inattendus conformément au § 312.32 ou omet de faire tout autre rapport requis en vertu de la présente partie.
· Les données du VAERS ont été délibérément et inconsciemment ignorées par la FDA, le CDC, le NIH et Pfizer. En outre, il existe des dizaines de milliers, voire des centaines de milliers de témoignages de victimes, de membres de la famille des victimes et de prestataires de soins de santé de l'expérience du « vaccin » EUA COVID-19, qui ont été ignorés ou rejetés par les agences gouvernementales impliquées et leurs collaborateurs des marchés des médias grand public, des médias sociaux, de la santé, des employeurs et de l'éducation.
RÉPONSE BLA DE LA FDA : ¶3:2-5
« … l'existence de rapports dans le VAERS ne signifie pas qu'une relation de cause à effet a été établie. La FDA a constaté que de nombreux rapports dans le VAERS ne documentent pas qu'un vaccin a causé l'événement signalé... Des rapports de décès après des vaccinations COVID-19 qui se révèlent être liés, ou même éventuellement liés, à la vaccination avec des vaccins COVID-19 ont été extrêmement rare." – Janet Woodcock, M.D.
Dr Woodcock, votre déclaration écrite concernant votre opinion ou vos déclarations selon lesquels les décès et les événements indésirables graves dans le VAERS liés aux vaccins COVID-19 est fallacieuse.
Selon la page 6, ¶3 de la lettre d'approbation FDA-Pfizer BioNTech BLA du 23 août 2021, la FDA déclare ;
"Nous avons déterminé qu'une analyse des événements indésirables post-commercialisation spontanés signalés en vertu de l'article 505 (k) (1) de la FDCA ne sera pas suffisante pour évaluer les risques graves connus de myocardite et de péricardite et identifier un risque grave inattendu de myocardite subclinique"
Le paragraphe ci-dessus fait spécifiquement référence aux données collectées dans VAERS, et que ces données sont suffisantes pour établir une corrélation entre le vaccin Pfizer BioNTech Covid-19 et la myocardite et la péricardite, en tout cas suffisamment pour justifier la réalisation de plusieurs études de phase 4 post-commercialisation afin de déterminer le degré, la gravité et les séquelles des maladies cardiaques causées par les «vaccins».
En outre, selon le document FDA/CBER d'octobre 2019, Postmarketing Studies and Clinical Trials —Implementation of Section 505(o)(3) of the Federal Food, Drug, and Cosmetic Act Guidance for Industry, indique clairement :
La détermination du …. l'autonomie du VAERS pour atteindre les objectifs décrits à l'article 505(o)(3)(B) de la loi FD&C est fondée, dans le contexte du risque grave lié à l'utilisation d'un médicament particulier, sur des considérations relatives aux forces et aux limites des rapports d'événements indésirables comme sources d'information et sur les caractéristiques particulières des données du …. Systèmes VAERS. Par conséquent, la FDA détermine …. la pertinence de données du VAERS aux fins d'exiger ou non une étude de post-commercialisation ou un essai clinique au cas par cas pour chaque risque grave.
Les documents et réglementations de votre propre administration stipulent clairement que les études d'innocuité post-commercialisation sont justifiées lorsqu'il existe une corrélation entre un événement indésirable grave et un médicament expérimental (vaccin).
DES QUESTIONS:
· Pourquoi n'êtes-vous pas d'accord avec les directives de votre propre administration ?
· Si les raisons de mener les études d'innocuité post-commercialisation pour la myocardite et la péricardite n'étaient pas fondées sur les preuves réelles de maladies cardiaques et de décès induits par le vaccin selon la base de données VAERS, à votre avis d'expert, sur quoi ces études post-commercialisation étaient-elles basées ?
Dr Woodcock, je pense que tous les Américains seraient d'accord pour dire qu'il y a eu beaucoup de confusion concernant les informations exactes sur ces vaccins, ce qui a accru les inquiétudes liées à leur sécurité. Par sec. 3024 du 21st Century CARES Act, le droit du peuple américain à un consentement éclairé en vertu d'un médicament ou d'un vaccin EUA n'est pas requis.
· (Sec. 3024) Les tests cliniques de dispositifs médicaux expérimentaux et de médicaments ne nécessitent plus le consentement éclairé des sujets si les tests ne présentent qu'un risque minimal pour les sujets et incluent des garanties.
Il y a trop de violations flagrantes des droits de l'homme permises par Sec. 3024 , y compris les crimes contre l'humanité, et dans l'intérêt de la productivité et de la transparence, je crois qu'après 18 mois d'obligations absurdes, avec confinements , port de masques, distanciation sociale, et maintenant l'émergence dangereusement agressive des obligations de vaccins, qu'au minimum, le peuple américain a le droit de savoir ce que contiennent les « vaccins » Pfizer BioNTech EUA et BLA COVID-19.
Le 7 juin 2021, AGC Biologics a annoncé des accords de fabrication pour produire de l'ADN plasmidique (ADNp) pour BioNTech, le partenaire de fabrication de Pfizer-BioNTech.
DES QUESTIONS:
· Quelle est la différence entre pDNA et rDNA ?
· Quel est son rôle de pDNA dans les vaccins à ARNm ?
Selon les dépôts de phase 3, le projet de notice d'emballage, PfizerBioNtech BNT162b et Moderna mRNA-1272 contiennent des nanoparticules lipidiques (LNP).
Selon le brevet mondial qui couvre les vaccins ARNm LNP, WO 2020/160397 A1, délivré à Moderna le 6 août 2020, couvrant «l'art» ou la propriété intellectuelle des vaccins Pfizer BNT162 et Moderna ARNm-1273, la Sec. 0002 indique :
· La présente invention concerne de nouveaux procédés de production de formulations de nanoparticules lipidiques (LNP) d'acide nucléique (« ARNm »)… et les utilisations thérapeutiques ou diagnostiques associées.
Un diagnostic n'est pas un vaccin. Un diagnostic est un dispositif médical.
En vertu du TITRE 21 de la loi FD&C, Sec. 814.9 : Confidentialité des données et informations d'approbation préalable à la mise sur le marché (PMA) des dispositifs médicaux, précise ;
(b) L'existence d'un dossier PMA ne peut être divulguée par la FDA avant qu'une ordonnance d'approbation ne soit délivrée au demandeur, à moins qu'elle n'ait été préalablement divulguée ou reconnue publiquement.
DES QUESTIONS:
· Quel(s) est(sont) le(s) dispositif(s) médical(aux) des « vaccins » Pfizer BioNTech ?
· De quels matériaux sont-ils composés ?
· Quelles sont les fonctionnalités des dispositifs médicaux en termes de transmission et d'échange de données ?
· Quels sont les risques associés de lésions permanentes, d'invalidité ou de décès pour les sujets humains auxquels ces dispositifs médicaux ont été injectés ?
En résumé, sur la base des informations disponibles, soumises et publiées par la FDA, il semble que les vaccins COVID-19 ne procurent aucun avantage clinique et ne peuvent que nuire, handicaper et tuer les Américains.
S'il existe des preuves cliniques et scientifiques qui réfutent clairement les affirmations ci-dessus, je vous prie de communiquer immédiatement ces données au peuple américain. Vous devez divulguer immédiatement au peuple américain toute information concernant les événements indésirables de ces vaccins et de leurs composants.
Les coloriages sont un choix de ce blog


Commentaires
Enregistrer un commentaire