Qu’est-ce qu’un « essai clinique » et qu’est-ce qui n’en est pas un ?
https://sashalatypova.substack.com/p/what-is-and-isnt-a-clinical-trial?

Je rédige actuellement une importante plainte. Je la publierai une fois terminée et déposée. Ce faisant, je me suis rendu compte que de nombreux termes utilisés couramment dans la publicité, la propagande et autres formes de manipulation de l'opinion publique sont mal définis. Bien sûr, c'est intentionnel. Voici donc un « résultat indirect » de mon travail en cours. J'espère qu'il sera utile à celles et ceux qui luttent contre ce complot d'empoisonneurs.
En droit américain, il n'existe pas de définition unique de l'« essai clinique ». Trois définitions, issues de trois cadres
juridiques différents, sont en vigueur. Ces définitions prêtent
facilement à confusion et, à mon avis, cette confusion a été exploitée
par les partenariats public-privé qui tirent profit des pouvoirs
conférés par l'« urgence pandémique ».
Les trois définitions juridiques :
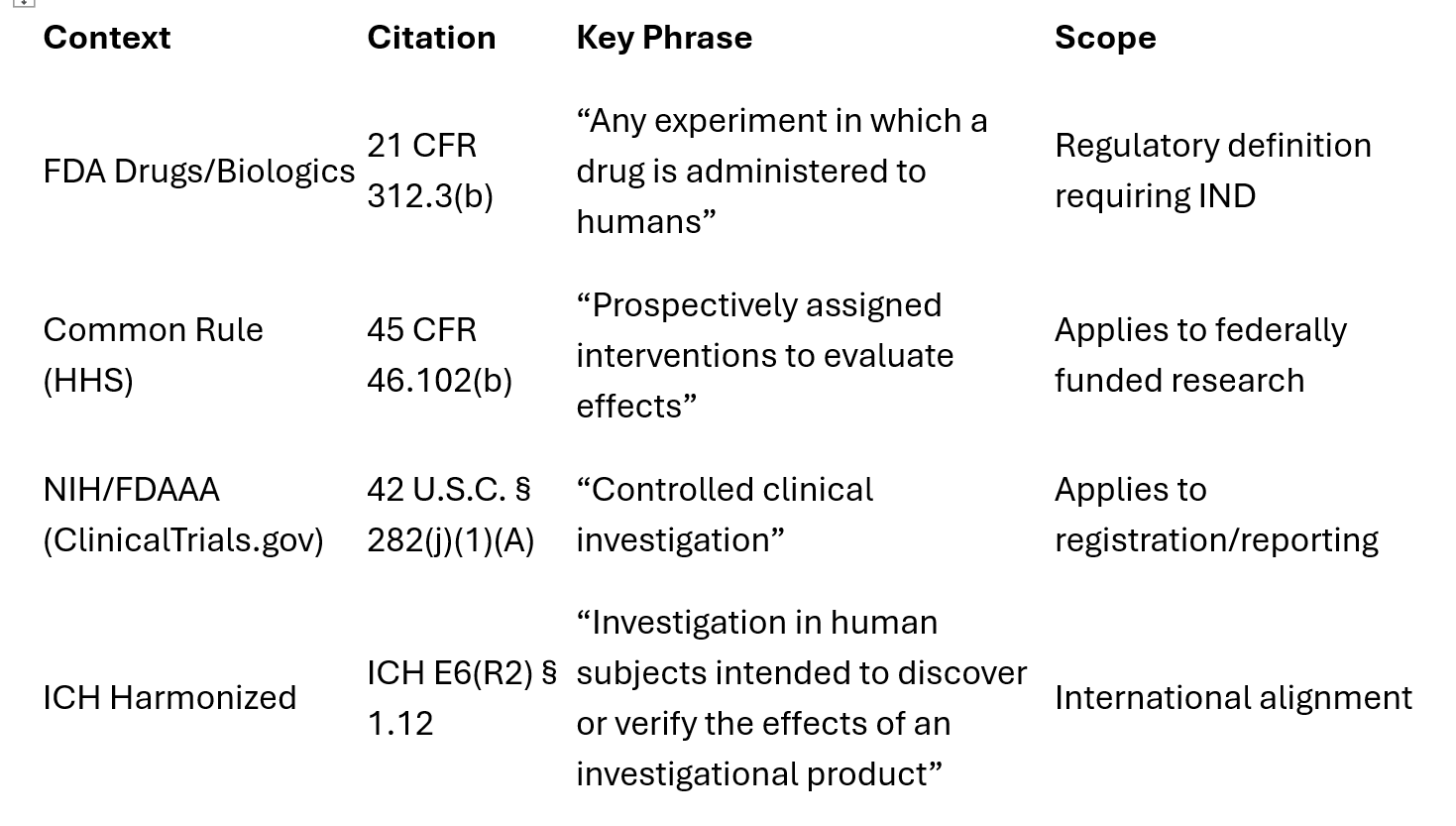
1) Cadre d’utilisation expérimentale, FDA (médicaments/produits biologiques) — 21 CFR Partie 312 :
La FDA n’utilise pas le terme « essai clinique ». Elle utilise « investigation clinique » (“clinical investigation”), définie comme suit :
« Toute expérience au cours de laquelle un médicament est utilisé sur des êtres humains, sauf lorsqu’un médicament commercialisé est utilisé « dans le cadre de la pratique médicale ». »
Cette définition de l'essai clinique correspond à ce que la plupart des gens entendent par essais cliniques : des études formelles de nouveaux médicaments et dispositifs médicaux en vue de leur approbation par la FDA et de leur autorisation de mise sur le marché. Ce cadre réglementaire impose au promoteur (fabricant) de déposer une demande d'autorisation de mise sur le marché d'un nouveau médicament expérimental (IND) et d'obtenir une exemption IND pour pouvoir entamer le processus d'essais cliniques.
Pourquoi une « exemption » ? Parce que les pouvoirs réglementaires de la FDA découlent de l'interdiction de commercialiser des médicaments, des produits biologiques et des dispositifs médicaux « non approuvés » dans le commerce interétatique. Elle utilise ce pouvoir, par exemple, pour s'en prendre à quiconque menace, selon elle, les grands groupes pharmaceutiques et hospitaliers avec lesquels elle entretient des liens douteux, y compris les vendeurs de lait cru et de compléments alimentaires, ou toute personne ayant découvert des méthodes non toxiques et efficaces pour traiter le cancer. C'est absolument inacceptable !
Pour
revenir au cadre de la demande d'autorisation de mise sur le marché
(IND), lorsqu'un promoteur souhaite obtenir l'approbation de la FDA, il
doit s'engager formellement à respecter la législation et la
réglementation relatives à la recherche clinique
. La procédure IND établit ce contrat formel entre le promoteur
(fabricant) du médicament et la FDA. Une fois ce contrat signé, le
promoteur est exempté de l'interdiction de commercialiser un produit non
approuvé, mais uniquement pour la réalisation d'une investigation
clinique réglementée, menée sur des sujets humains et conforme à la loi.
La supervision des essais cliniques est assurée par le comité d'éthique
de la recherche et les règles relatives au consentement éclairé (21
CFR, parties 50 et 56, et loi nationale sur la recherche ).
2) La règle commune (HHS)(The Common Rule) — 45 CFR 46.102(b)
Le cadre de la « règle commune » s’applique plus largement à la recherche financée par le gouvernement américain, notamment à la recherche menée par les établissements universitaires et les fabricants commerciaux, mais qui ne fait pas partie du processus formel d’approbation des médicaments ou des dispositifs médicaux. Elle porte sur l’attribution prospective et l’évaluation des résultats de santé, et non pas seulement sur l’utilisation des médicaments.
Dans ce contexte, le terme « essai clinique » désigne une étude de recherche dans laquelle un ou plusieurs sujets humains sont assignés de manière prospective à des interventions (y compris un placebo) afin d’évaluer leurs effets sur des critères d’évaluation biomédicaux ou liés à la santé comportementale.
Comme
dans le cadre d’un essai expérimental, l’examen par un comité d’éthique
de la recherche et le consentement éclairé sont obligatoires et régis
par les mêmes lois.
3) FDAAA/NIH ( ClinicalTrials.gov ) — 42 USC § 282(j)
Cette définition est utilisée aux fins de l'enregistrement des essais cliniques et de la communication des résultats :
« une étude clinique contrôlée autre que la phase 1 d’un médicament/produit biologique soumis à la loi FDCA/PHS ».
Cette
définition est plus restrictive que celle de la partie 312 ou de la
Common Rule. De nombreuses études sont des essais cliniques relevant de
ces régimes, mais ne constituent pas des « essais cliniques
applicables » soumis à l’obligation d’enregistrement et de déclaration.
En plus de ces définitions, il existe un quatrième cadre (Conférence internationale sur l'harmonisation, ICH) qui est utilisé pour les programmes mondiaux/internationaux, mais il ne s'agit pas du droit américain.
En résumé, voici l’ensemble des cadres juridiques applicables aux essais cliniques. Il convient de noter que tous utilisent le terme « expérimental » comme mot-clé dans leurs définitions.
Pour bien définir quelque chose, il faut aussi examiner ce qu'il n'est PAS.
Qu’est-ce qui n’est pas un essai clinique (même si quelqu’un le présente comme tel) ?
Plusieurs utilisations de produits médicaux peuvent être qualifiées, de manière informelle, d’« essai clinique », mais ne sont pas considérées comme telles au regard des normes juridiques. Ces utilisations ne constituent pas des essais cliniques, mais des voies d’accès au traitement. La législation américaine identifie également les catégories suivantes, particulièrement importantes en cas de litiges :
1) Pratique médicale avec un produit commercialisé :
Le traitement d’un patient avec un médicament ou un dispositif médical
déjà approuvé par la FDA ou commercialisé légalement – même hors
indication – est généralement considéré comme un acte médical et non
comme une recherche clinique. Pour les dispositifs médicaux, le Congrès
américain l’a explicitement indiqué au titre 21 du Code des États-Unis,
section 396 ; la FDA applique le même principe aux médicaments dans ses
politiques et son application.
Si
le produit est utilisé pour traiter un seul patient dans le cadre de
soins personnalisés, il relève de la pratique médicale. En revanche,
s'il est utilisé pour recueillir des données auprès de plusieurs sujets
dans le cadre d'une étude prospective avec un plan d'analyse, cette
activité pourrait relever de l'« essai clinique » et nécessiter
l'approbation d'un comité d'éthique de la recherche et le consentement
éclairé des participants.
2) Accès aux produits expérimentaux hors essais cliniques :
Nous revenons ici à la notion de produits « expérimentaux ». Plusieurs
voies permettent d’utiliser des traitements médicaux en dehors de la
recherche clinique :
Accès élargi (« usage compassionnel ») — utilisation du traitement conformément à la partie 312, sous-partie I, du titre 21 du CFR . L’objectif est de prodiguer des soins, et non de tester des hypothèses. L’examen par un comité d’éthique de la recherche demeure obligatoire.
Utilisation d’urgence (patient unique, urgente) — autorisée une fois sans approbation préalable du comité d’éthique de la recherche, avec notification au comité d’éthique de la recherche dans les 5 jours ouvrables (21 CFR 56.104(c)).
3) Accès hors essais cliniques à des produits NON expérimentaux :
Autorisation d'utilisation d'urgence (EUA) des contre-mesures. Conformément à l'article 360bbb-3 du titre 21 du code des États-Unis, définie comme
Autorisation temporaire pour des produits non homologués ou des utilisations non homologuées de produits homologués pendant une urgence de santé publique déclarée.
Il s'agit d'une autorisation temporaire à l'échelle de la population, accordée dans le cadre d'une situation d'urgence déclarée. La loi visait officiellement le déploiement, par les autorités de santé publique, de contre-mesures face aux attaques à l'arme NRBC. Cependant, le Département de la Santé et des Services sociaux (HHS) affirme aujourd'hui qu'elle s'applique également aux « agents pathogènes émergents », une définition vague.
Quoi qu'il en soit, cette utilisation des produits n'est pas juridiquement définie comme de la « recherche clinique » et ne peut donc être considérée comme un « essai clinique ».
Dans ce contexte, les produits médicaux ne sont plus considérés comme tels, mais comme de simples « contre-mesures ». Leurs caractéristiques physiques ou chimiques, leurs propriétés thérapeutiques, leur innocuité ou leur efficacité, ou encore leur statut antérieur à l'autorisation d'utilisation d'urgence (EUA) (« approuvé par la FDA » ou « non approuvé »), deviennent sans objet.
En effet, ces propriétés et attributs ne sont juridiquement définis que pour les produits « expérimentaux », soumis à la législation sur la recherche clinique avec consentement éclairé et soumis à l'approbation d'un comité d'éthique de la recherche (IRB), et destinés à un usage médical.
Autrement dit, si l'objectif est d'utiliser un appareil à rayons X comme presse-papier, son approbation par la FDA pour l'imagerie médicale diagnostique est totalement insignifiante.
Si l'intention est de larguer la même machine du 20e étage sur la tête d'un ennemi (une « contre-mesure »), il s'agit également d'une utilisation non expérimentale et non médicale de ce dispositif, non soumise aux normes de sécurité et d'efficacité de la FDA. Le fait que la machine larguée du 20e étage ait été fabriquée dans le strict respect des normes cGMP est également sans incidence juridique, et il est impossible d'obliger la FDA à retirer ce dispositif du marché sous prétexte qu'il serait « dangereux ». Les fondements juridiques que constituent l'approbation d'un comité d'éthique de la recherche (IRB) et le consentement éclairé sont explicitement exclus de ce type d'utilisation d'un produit médical.
De plus, toutes les protections et exigences réglementaires normalement applicables en matière de sécurité des consommateurs sont levées ou rendues inapplicables pour les utilisations d'une unité d'autorisation de mise sur le marché (EUA), et les fabricants bénéficient d'une protection juridique absolue grâce à la loi PREP et autres textes législatifs connexes.
J'ai traité en détail des dispositions légales qui
suppriment toute protection des consommateurs pour les EUA dans cet article , et j'ai publié une introduction complète à la loi PREP dans cet article .
Par ailleurs, comme l’explique la FDA dans son guide intitulé « Autorisation d’utilisation d’urgence des produits médicaux et autorisations connexes » (révisé en 2024) :
« Étant donné que l’administration dans le cadre d’une EUA n’est pas une investigation clinique , les exigences d’examen par un IRB et de consentement éclairé en vertu des parties 50 et 56 du titre 21 du CFR ne s’appliquent pas . »
— Directives de la FDA, Autorisation d’utilisation d’urgence des produits médicaux et autorisations connexes (2024), Section III.C.5.
Au lieu du consentement éclairé juridiquement valable, les destinataires des contre-mesures n’ont droit qu’à une simple « fiche d’information » :
«
Les destinataires doivent être informés, dans la mesure du possible,
des avantages et des risques importants, connus et potentiels, du
produit et de la possibilité de l’accepter ou de le refuser. »
— Id., § VI.C.3.
En conclusion – la frontière légale :
La limite légale entre ce qui constitue un « véritable » essai clinique et ce qui ne l’est pas réside dans la finalité et l’intention de l’utilisation spécifique du produit. La FDA distingue ces utilisations non expérimentales de la recherche clinique pour les raisons suivantes :
« L’objectif de l’accès élargi est de traiter un patient, et non d’obtenir des informations sur l’innocuité ou l’efficacité d’un médicament. »
— 21 CFR § 312.300(b)
Par conséquent, aucune de ces voies — accès élargi, utilisation d’urgence ou EUA — ne constitue un essai clinique ou une investigation clinique au sens du § 312.3(b).
Œuvre d'art du jour : Trois pivoines, huile sur panneau, 9x12 pouces .


Un texte des hospices civils de Lyon expose les diverses questions que pourrait soulever l'expression "essai clinique" https://www.chu-lyon.fr/essai-clinique
RépondreSupprimerVous noterez à l'étude la phase III la comparaison éventuelle à faire avec un placebo. MÊÊÊÊ dans la 2ème partie de l'explication, le PLACEBO a déjà disparu. La comparaison NE SE FAIT PLUS qu'avec le traitement antérieur , dit, traitement de référence.
Et OUALA